Given the soaring costs of prescription drugs, it is not surprising that more than 70 percent of Americans think that drug prices are “unreasonable” and that “drug companies put profits before people.” The pharmaceutical industry and its allies have responded in part by trying to shift blame to the federal Food and Drug Administration, or FDA, claiming that the reason drug prices remain high is because the FDA approval process is too burdensome and slow, keeping competing drugs from the market and “stifling American innovation.”
This argument is not just misleading, it is dangerous. The drug approval process needs to strike a careful balance between speed and diligence—patients need safe, effective drugs, and it takes time and clinical trials in order to determine whether a drug meets those standards. Today, the FDA approves the overwhelming majority of new drugs, and does so at a quicker pace than any other nation. Furthermore, federal law already allows the FDA to shorten both the clinical trial and approval processes for a large number of drugs in order to speed them to market. Yet even for drugs deemed more innovative or urgently needed, trade-offs persist; drugs approved under these expedited programs can later be found to have dangerous side effects or be far less effective than first thought.
Despite these risks and the FDA’s existing authority to expedite review for new treatments in order to meet patient need, the pharmaceutical industry continues to push for policies that are designed to hasten drug approvals. For example, the 21st Century Cures Act includes a number of provisions aimed at expanding the types of data that the FDA may rely on when determining whether a drug is safe and effective. Since these additional types of data are generally less rigorous, this would lead to more drugs entering the market with far less information about their risks and evidence about their potential benefits.
Even if there were no concerns that these reforms increase risks to patients, there is little reason to believe that pushing new branded drugs to market would guarantee meaningful price reductions in the absence of generic competition. Newly approved drugs enjoy patent and marketing exclusivity that limit competition, and manufacturers continue to set prices based on what they think the market will bear, not the drug’s value to patients.
This issue brief first will describe how drugs are developed and approved in the United States. It then will explore the reasons why undermining the FDA’s authority and speeding up the drug development and approval process will harm patients and do nothing to lower drug prices.
Drug development and approval processes
Today, the FDA’s approval process is, on average, faster than that of any other major nation. The FDA is faster than both the European Union and Japan not only in the overall average approval time for new drugs but also in the average time for every major category of drug. Similarly, studies comparing the FDA with Canadian drug regulators have found that the FDA consistently approves drugs more quickly. In 2015, 64 percent of innovative new drugs were approved by the FDA before they received approval in any other country in the world.
In response, the pharmaceutical industry and its allies point out that the actual regulatory review time is just one part of the drug development and approval process. Some critics argue that the pre-FDA process of drug development and testing—including the various stages of clinical trials—takes too long and costs too much, thus stifling innovation.
It is true that bringing a new prescription drug to market is a time-consuming, expensive, and risky process for drug manufacturers. But the FDA approval requirements exist to ensure that new drugs are safe and to prove that they actually work. The current U.S. drug approval process has evolved as policymakers have weighed competing concerns—including the significant financial investment that drug manufacturers make throughout the clinical trial and approval processes, the need for drugs to be safe and effective, and the need to speed a drug to market in cases where there are gaps in treatments.
Standard drug development and the FDA approval process
For all drugs, the pharmaceutical industry generally uses the findings from basic research, which studies the mechanisms of diseases, as a starting point for its applied research and development efforts. Basic research is funded in large part by the federal government and conducted by researchers at the National Institutes of Health and in academic laboratories.
The first step of industry-sponsored research is laboratory and animal testing to evaluate if the investigational use of the new product in people is reasonably safe. The FDA reviews these findings, and if it agrees that the results show that the product is reasonably safe for people, the company may move forward with clinical trials, which test the drug in patients.
These clinical trials occur in several phases, with each phase testing the drug in increasingly larger groups of patients in order to gather information about the product’s safety and effectiveness, as well as side effects, dosing, and interactions with other drugs, food, and drinks. Phase 1 studies, which typically consist of 20 to 80 healthy volunteers, study the drug’s toxicity and safety, as well as how people process the drug. If the drug is not unacceptably toxic, Phase 2 studies follow. These studies start to consider the drug’s effectiveness by collecting preliminary data on how the drug works in patients with specific diseases or conditions, while continuing to study the drug’s safety and side effects. The number of people enrolled in a Phase 2 study can range from a few dozen to about 300.
If the Phase 2 studies show evidence of effectiveness, the drug manufacturer and the FDA will try to agree to the design of the Phase 3 study. The FDA monitors clinical trials throughout their entire duration, but the pre-Phase 3 period is one of the most common meeting points throughout the clinical trial process. Phase 3 studies collect additional information about safety and effectiveness and can study the drug’s effect in different populations, at different dosages, and when combined with other drugs. These studies typically range in size from several hundred to about 3,000 people.
When the company believes that these studies offer enough evidence to show that the product is safe and effective, it will seek the FDA’s approval by submitting a New Drug Application, or NDA, that includes all preclinical and clinical trial data, as well as information about how the drug works in the body and how it is manufactured. It is also common for manufacturers and the FDA to meet at this point, right before an NDA is submitted.
After the NDA submission, the FDA has 60 days to decide whether the application is complete and ready for review. The Prescription Drug User Fee Act, or PDUFA sets review timelines for the FDA, and the FDA’s goal is to review and act on at least 90 percent of NDAs for drugs under the standard review process no later than 10 months after the applications are received.
The FDA approves a drug for marketing and sale if the data show that the drug is safe and effective in its proposed use, and if its benefits outweigh the known risks. Federal law requires “substantial evidence” that the drug is safe and effective. Substantial evidence means, in part, “adequate and well-controlled investigations, including clinical trials.” As FDA explains, the “NDA is supposed to tell the drug’s whole story, including what happened during the clinical tests, what the ingredients of the drug are, the results of the animal studies, how the drug behaves in the body, and how it is manufactured, processed and packaged.” As a condition of approval, the FDA may require the manufacturer to conduct additional postmarket studies to continue to gather data about the drug’s safety and effectiveness.
Expedited programs
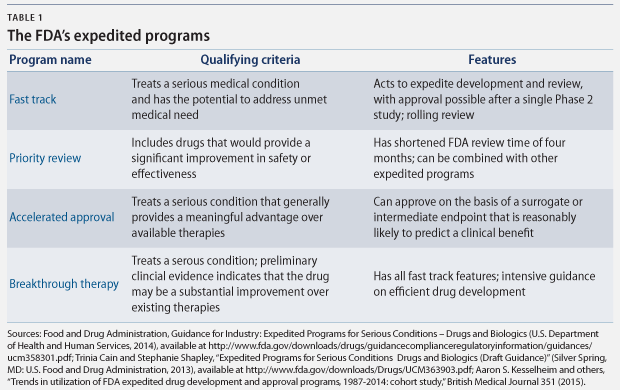
There are a number of FDA programs that policymakers have designed to speed the development and approval of new drugs that “address unmet medical need in the treatment of a serious or life-threatening condition.” The four principal programs are fast track designation, breakthrough therapy designation, accelerated approval, and priority review. Each program expedites drug approvals for serious conditions, but each has different qualifying criteria and approval requirements. (see Table 1)
According to the FDA, these pathways “help ensure that therapies for serious conditions are approved and available to patients as soon as it can be concluded that the therapies’ benefits justify their risks.” These pathways are of particular importance when there are few—or no—treatment options, and they are particularly helpful “in settings in which the disease course is long and an extended period of time would be required to measure the intended clinical benefits of a drug.” Between 2000 and 2013, 32 percent of new molecular products, including biologics, which are drugs made from living cells, were approved under the accelerated approval and fast track pathways.
Although the FDA’s standard review time is about 12 months, these programs speed up the review process. More importantly, however, the drug development process can be shortened in three of these four programs, because the FDA can make its determination of the drug’s safety and effectiveness based on limited clinical data. Shortening the clinical trial period speeds critical new drugs to market, but it comes with a risk, because there is limited data to prove their safety and effectiveness.
Under the fast track designation, for example, the FDA may approve a drug based on data from a single Phase 2 study. And the accelerated approval pathway allows approval based on surrogate endpoints that are reasonably likely to predict patient outcomes. Surrogate endpoints are markers such as laboratory results or radiology images, while clinical endpoints measure the reduction in symptoms or mortality. For cancer treatments, surrogate endpoints used to approve drugs include a shrinking tumor or lower biomarker levels, instead of the clinical endpoints of longer survival or improved quality of life.
Surrogate endpoints can be measured sooner, allowing patients access to new treatments much faster, but they are not always accurate indicators of how well a treatment may work, especially in a larger real world population. As one study noted, “drugs that are approved after a shortened premarket period or based on … [limited data] may later be found to have greater risks or less certain benefits than initially believed to be the case.” For this reason, drugs approved using limited data are then subject to postapproval testing to ultimately confirm that they are in fact safe and effective.
Drug approval rate
Under the current regulatory structure, the FDA approves almost every new drug application it receives. In 2015 and 2014, the FDA approved 89 percent and 100 percent, respectively, of novel drug applications, which are defined as more innovative NDAs that involve new molecular entities. While not unprecedented, this is a higher approval rate than the FDA generally has produced in the past; in 2007 and 2008, for example, the FDA approved 51 percent and 71 percent of novel drug applications, respectively.
Other sources confirm the FDA’s numbers. Forbes commissioned an analysis of FDA approvals from BioMedTracker and concluded that “the FDA is basically approving everything.” In other words, not only are the FDA’s standards not overly harsh, they are currently unusually lenient.
Undermining the FDA approval process will benefit the drug industry, not patients
Given the speed and approval rate of the FDA, there is no need to further expedite the drug approval process. This approach would lead to additional drugs entering the market with little evidence to support their safety and effectiveness, which can harm patients. In fact, growing numbers of experts—including a former FDA commissioner—have warned that weakening the FDA’s approval standards by changing the types of evidence that the FDA reviews when approving drugs would put patients at risk.
Nevertheless, many lawmakers support the 21st Century Cures Act and other similar proposals. As noted previously, the 21st Century Cures Act includes a number of troubling policies that would give FDA the authority to find a drug safe and effective on the basis of less scientifically rigorous evidence.
For example, the legislation lays the groundwork for FDA to use broader categories of evidence related to “clinical experience,” which includes “observational studies, registries, and therapeutic use” instead of randomized controlled trials for approving new uses for existing drugs or to satisfy post-approval study requirements. But as experts caution, “although such data can provide important information about drug utilization and safety once a medication is in use, there is considerable evidence that these approaches are not as rigorous or valid as randomized trials in assessing efficacy.” Another section of the bill authorizes the FDA to approve new antibiotics and antifungal medicines intended to treat serious or life-threatening infections in certain patients based on limited data, including preclinical trials. Moreover, the legislation creates a financial incentive for hospitals to use these largely untested drugs: For each patient treated with these newly approved drugs, the hospital receives an add-on payment.
Despite the significant limitations of surrogate endpoints, the legislation also encourages the FDA to expand its use of them to assess a drug’s safety and effectiveness. The FDA, however, already has the authority to use these data when it is most appropriate: when the drug is intended to treat a serious or life-threatening condition. It also gives industry experts an increased role in determining when it is appropriate to use these surrogate endpoints.
Ultimately, however, there is no evidence that changing these standards would speed the approval of game-changing, truly innovative new products, nor is there any evidence that less rigorous review would lower drug prices.
Approving drugs with less evidence would put patients at risk
The current expedited programs already place a premium on speed of approval. When drugs are approved using an expedited pathway, it comes with a trade-off: less information and data about how the drug acts in a patient’s body.
In situations where there are few existing treatments or where a new treatment can significantly improve patient outcomes, speed is a priority. But if the drugs being approved are less urgently needed or offer little to no improvement over existing treatments, the need to prioritize speed over safety diminishes. In such cases, the benefits are much less likely to outweigh the risks to patient safety that are inherent in permitting drugs to enter the market after shortened clinical trials or based on limited data.
Evidence supports this concern. For example, researchers have found that since 1992, when policymakers adopted the priority review and accelerated approval programs, the number of approved prescription drugs that received black-box warnings—which is the most serious safety warning that the FDA can impose on a drug—or that were withdrawn from the market for safety-related reasons has increased 25 percent. The researchers suggest that one reason for this increase is that growing numbers of new drugs are entering the market with more limited data about their safety and efficacy.
Notably, the Government Accountability Office has already raised concerns that the FDA lacks sufficient data to thoroughly monitor the safety of new drugs after they have been approved—especially for drugs that are approved under an expedited pathway, where postapproval oversight is crucial due to the abbreviated clinical trial process.
This worry will become even more significant in the coming years, because an increasing number of drugs are being channeled into expedited programs. In 2014, more than 60 percent of approved drugs were approved by the FDA based on reduced data and evidence requirements. The FDA now approves more than two-thirds of drugs based on data from studies lasting six months or less. Moreover, one-third of new drugs are approved on the basis of a single trial, and the median size for all such trials is just more than 750 patients. The FDA is also approving an increasing number of supplemental new drug applications for additional clinical uses or patient populations—including children—using surrogate endpoints and limited data. Some of these additional uses might be truly innovative for the newly approved uses or patient groups and allow drug companies to market these drugs to populations in need of new treatment options; as the study’s authors note, however, these findings once again demonstrate “the importance of post-approval surveillance of drugs’ supplemental indications, particularly those that expand the eligible patient population.”
Current FDA requirements do not stifle innovation
Under today’s expedited programs, truly transformative drugs can reach patients quickly. Gleevec, which has been described as a “miracle drug” that can turn deadly cancers into chronic conditions, is an excellent example of how well this process can work. Gleevec was first approved in 2001 for the treatment of chronic myelogenous leukemia—a rare blood cancer—after only Phase 2 studies and just 2.5 months of FDA review. Since its initial approval, the FDA has approved additional uses of the drug to treat several different gastrointestinal tumors, and post-marketing studies have confirmed its effectiveness.
Gleevec’s approval shows how the FDA can act quickly to approve truly transformative products. But not all drugs that go through expedited programs are similar game-changers. A study by researchers at Harvard Medical School and Brigham and Women’s Hospital found that from 1987 to 2013, the number of drugs that the FDA approved under expedited programs increased 2.6 percent each year. Yet drugs that were not “first in class” drove this trend – meaning the drugs followed functionally similar products. The study authors concluded that although “some drugs associated with an expedited program may indeed provide noticeable clinical advances, this trend is being driven by drugs that are not first in class and thus potentially less innovative.”
Skewing the process toward greater speed is simply not necessary. And it is illogical to think that such changes will necessarily lead to future breakthroughs like Gleevec. On the contrary, Gleevec shows that the current process works to expedite innovative drugs; efforts to allow more drugs on the market with even lower evidence of safety and effectiveness will put patients at risk unnecessarily.
Speeding drugs to market will not necessarily lower prices
Proponents of loosening the FDA’s requirements often argue that speeding the drug approval process will help reduce prices. Yet there is little indication that past policy changes to the FDA have had this effect; at the same time as the FDA has approved an increasing number of drugs through expedited pathways, drug prices have continued to rise.
Part of the tension in this argument is that truly breakthrough products that represent significant advances or address urgent medical needs—the precise drugs that should be approved through expedited pathways based on more limited information—generally by definition have no real competition or price constraints after entering the market. Once again, Gleevec is a good example of the disconnect between expedited approvals and price. When the drug was first approved, its manufacturer, Novartis, charged about $4,500 in 2014 dollars for one month of treatment. In 2014, that price had gone up to nearly $8,500 per month.
It is true that speeding competitor drugs to market can help reduce prices in some cases. Branded competition can give payers additional leverage to negotiate sizable discounts or limit price increases by tying drug coverage to price. For example, insurers can encourage demand for a specific drug by lowering its cost-sharing in exchange for price discounts from the manufacturer. But each situation differs based on the similarity of the competing drugs, the pricing strategies of the drug companies, and other available treatment options.
A recent example is that of high-priced new cures for Hepatitis C, when Viekira Pak followed Sovaldi to market only a year later. The pharmacy benefit manager Express Scripts was able to negotiate a large discount by making a deal with Viekira Pak’s manufacturer, AbbVie, to stop covering Sovaldi and exclusively cover Viekira Pak once the second drug hit the market. However, the enormous amount of media attention this received demonstrates the relative rarity of such high-profile arrangements. And although Express Scripts managed to receive a significant discount, Viekira Pak’s list price still closely tracked that of Sovaldi—demonstrating that branded competition does not necessarily correct for an excessive original price, since the original price often sets the price starting point for future competitors.
Notably, however, Sovaldi and its competitors received expedited approval from the FDA, which demonstrated that the agency can already act quickly to both expedite breakthroughs and help increase competition. The FDA is not the barrier in such situations; how quickly branded competition can appear depends primarily on whether comparable drugs are already in the development pipeline. Furthermore, the FDA later required AbbVie to add information to its label about serious liver injury risk with Viekira Pak*—demonstrating once again the trade-offs between speed and patient safety, even for innovative cures.
Yet, while interesting, the Sovaldi example is not representative of all drugs. Hepatitis C was an unusual situation in which an extremely high-priced breakthrough drug was followed relatively quickly by multiple competitors that had been close behind the original in the development process. Furthermore, outrage over the high price of the original drug, Sovaldi, opened opportunities for competitors to strike exclusive deals with major payers who were growing desperate.
For every example of a new drug spurring price competition, there is another of pharmaceutical companies taking advantage of shifting market dynamics in order to price their competitor drugs higher than the original drugs. Studies have found that in the absence of generic competition, increases in branded competition often only result in marginal price reductions—or as the following examples illustrate, even price increases.
For instance, although 11 major drug alternatives to treat multiple sclerosis have entered the market over the past two decades, all of them are priced in roughly the same high-cost range. These manufacturers have not attempted to undercut each other’s prices in order to gain market share. Rather, a study looking at nine of these drugs found that each time a new drug alternative has entered the market with a higher price, the manufacturers of the older drugs have raised their prices to match the new drug’s price. Four of the oldest drugs were originally priced 230 percent to 380 percent lower than they are now.
This stunning “shadow pricing,” in which drug competitors mirror each other’s price increases, also exists for other drugs. The prices for biologic rheumatoid arthritis drugs have all risen significantly as a group over the past few years. For example, the manufacturers of two rheumatoid arthritis drugs, Enbrel and Humira, both raised their prices in parallel by similar double-digit percentages in 2014 and 2013. Similarly, the insulin market has seen competitors raising their prices in tandem, often within days of each other; as a result, multiple brands of insulin saw price increases of 160 percent to 400 percent between 2007 and 2014.
In situations like these, monopoly protection and the manufacturers’ knowledge that they face no external limits on price appear to scramble the normal market calculations, with manufacturers raising prices to maximize profits as much as possible before they lose their marketing exclusivity and face generic competition. Essentially, manufacturers are aware that demand for their drugs is relatively inelastic: If they all stick together on high prices, patients who need these drugs will have nowhere else to turn.
This is not normal market behavior. And it should make clear that there is no normal, functioning market for prescription drugs. As a result, increasing branded competition alone will not guarantee price reductions.
There is no evidence that shortening the clinical trial or drug approval process will change these pharmaceutical industry behaviors. And while prices continue to rise, patients will be put at risk by the approval of drugs based on limited evidence.
The generic backlog
Critics of the FDA commonly point to the agency’s backlog of generic drug applications as their primary example of how the agency allegedly holds back competition. Yet while the backlog is a problem for generic drugs, no such backlog exists for brand name drugs. Often, the FDA’s detractors conflate these two separate issues, which overstates the impact of the backlog on drug approval and price inflation. Although generic drug price spikes harm patients and must be addressed, they have not been widespread enough to be a central contributor to recent increases in national drug spending; a recent analysis by the U.S. Department of Health and Human Services concluded that “they exert no sizable influence on overall drug spending.”
Furthermore, evidence suggests that the backlog is not even the primary factor in generic drug price inflation. Overall, 10 percent of all drugs have expired market exclusivity but lack generic drug applications submitted to the FDA, compared with the 2 percent that have submitted applications and are awaiting FDA approval. In other words, for more than 80 percent of off-patent drugs that lack generic competition, no generic manufacturer has submitted an application to the FDA. This suggests that lack of competition in the generic drug market—which leads to monopolies and price hikes—is primarily due to market dynamics that are unrelated to the FDA, such as mergers of generic manufacturers, drug shortages resulting from supply chain issues, and efforts to limit distribution through specialty pharmacies.
That being said, the FDA’s backlog of new generic drug applications is a significant problem that has existed for several years. In 2012, the Generic Drug User Fee Amendments, or GDUFA, authorized the FDA to collect fees from generic drug manufacturers, in part to finance an acceleration of the generic approval process. Yet despite the GDUFA, the backlog continues today, numbering roughly 4,300 at the end of 2015. Although most of the applications from the original backlog have been cleared by now, the volume of new applications over the past two years has been much higher than anticipated. The estimates used when developing GDUFA—and providing funding for the FDA’s Office of Generic Drugs—were for 750 new generic drug applications annually. In practice, however, the actual volume of applications since 2012 has dwarfed these estimates, leaving the Office of Generic Drugs underfunded and understaffed to process them. The FDA received more than 1,400 new generic drug applications in 2014 and received around 1,000 in both 2012 and 2013.
The FDA does use some criteria to prioritize applications in order to help manage the backlog, including whether an application is a first generic or whether a drug shortage exists. However, the FDA is not currently prioritizing based on the prices of existing drugs, or on whether those prices are increasing faster than inflation. Prioritizing on these factors could help maximize the impact of limited resources and reduce the potential impact of the backlog on generic drug prices.
Conclusion
Today, there is enough flexibility in the FDA approval process to bring lifesaving drugs to market in a timely process. And if policymakers are willing to increase funding for the FDA to review drug applications more quickly, more drugs could enter the market after appropriate vetting. Cutting corners by pushing the FDA to approve more drugs on the basis of more limited and less rigorous data, however, will simply put more patients at risk and do nothing to lower drug prices.
Maura Calsyn is the Director of Health Policy at the Center for American Progress. Thomas Huelskoetter is the Research Associate for Health Policy at the Center.
* Correction, March 11, 2016: This issue brief incorrectly stated the type of label change required by the FDA. To clarify, the FDA issued a safety announcement and required the manufacturer of Viekira Pak to add information to its label warning that the drug can cause serious liver injury mostly in patients with underlying advanced liver disease.